Common Problems in Cell Staining and How to Fix Them
Cell staining is one of the most widely used approaches in fluorescence-based cell analysis because it turns otherwise difficult-to-see structures, compartments, and cellular states into readable optical signals. But in practice, successful staining depends on far more than adding a dye and taking an image. Sample condition, dye chemistry, buffer composition, incubation time, washing stringency, fixation strategy, and acquisition settings all influence whether the final signal is specific, interpretable, and reproducible. When these variables are not aligned, cell staining can generate high background, weak fluorescence, uneven labeling, misleading co-localization, or signal changes that are incorrectly interpreted as biology. That is why troubleshooting cell staining should never begin with a single assumption such as "the dye concentration must be wrong" or "the microscope settings are bad." Most staining failures are multi-factorial. A weak signal may be caused by poor target accessibility, but it may also reflect photobleaching, poor sample health, or a dye that is fundamentally mismatched to the workflow. High background may arise from free dye residue, autofluorescence, non-specific staining, or a fixation-related artifact. The most effective way to solve these problems is to understand how the full workflow behaves from sample preparation to readout.
What Is Cell Staining?
Cell staining is the use of fluorescent dyes, fluorescent probes, or labeled reagents to convert otherwise difficult-to-observe cellular features into measurable signals. Those features may be structural, such as the nucleus, plasma membrane, cytoskeleton, or intracellular organelles, but they may also be functional, such as membrane integrity, intracellular transport, metabolic activity, ion-associated changes, or other state-dependent cellular properties. In fluorescence-based workflows, cell staining is therefore not just a method for making cells "visible." It is a strategy for extracting biologically meaningful information from a sample by linking a chemical reagent to a defined cellular target, compartment, or condition.
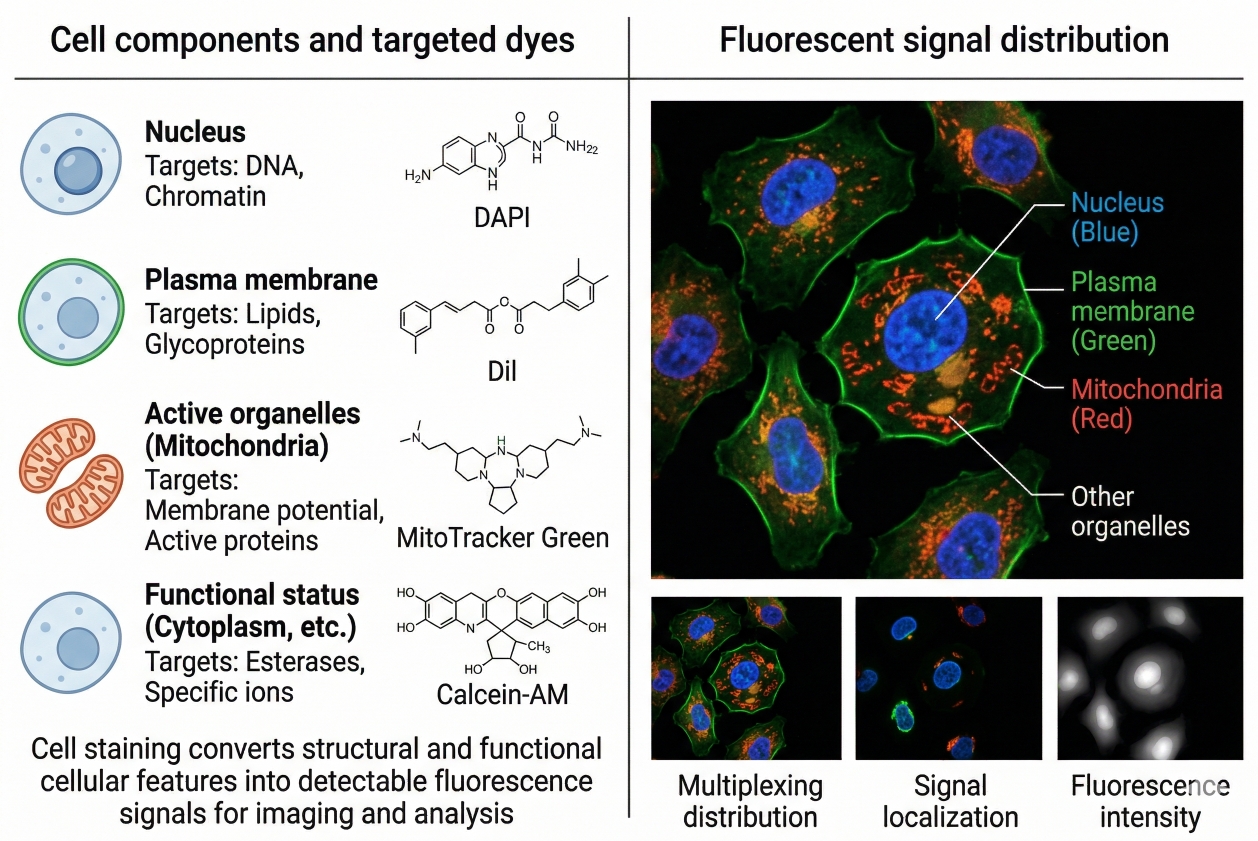 Fig. 1. Cell staining converts structural and functional cellular features into detectable fluorescence signals for imaging and analysis (BOC Sciences Authorized).
Fig. 1. Cell staining converts structural and functional cellular features into detectable fluorescence signals for imaging and analysis (BOC Sciences Authorized).
This distinction matters because cell staining is often discussed too simply, as though all stains operate in the same way and all fluorescent signals mean the same thing. In reality, staining methods differ profoundly in mechanism. Some dyes bind directly to a target structure, some accumulate in a compartment because of membrane permeability or electrochemical properties, some become fluorescent only after intracellular conversion, and some serve as general contrast agents rather than highly selective reporters. As a result, a staining outcome can only be interpreted correctly if the underlying staining logic is understood. A strong signal is not automatically a good signal, and a bright image is not automatically a meaningful one. Whether the fluorescence is useful depends on what the dye is designed to report and whether the experimental workflow preserves that meaning.
Structural Staining vs Functional Staining
One of the most important distinctions in cell staining is the difference between structural staining and functional staining. Structural staining is mainly used to show where a cellular feature is located, what shape it has, or how it is organized relative to the rest of the cell. Nuclear stains, membrane stains, cytoskeletal labels, and many organelle-directed reagents are often used this way. Functional staining, by contrast, is intended to report on a biological property or cellular condition rather than a static location alone. Viability dyes, membrane-potential-sensitive fluorophores, uptake probes, and other environment-responsive stains fall into this category. This distinction is critical in troubleshooting because a change in a structural stain often suggests altered localization, accessibility, or retention, whereas a change in a functional stain may reflect either a technical problem or a true biological shift in the state being measured. Without that distinction, researchers can misread technical artifacts as biology or ignore real biology as if it were staining failure.
Why Cell Staining Is Fundamental to Fluorescence-Based Cell Analysis?
Cell staining is fundamental because many of the most important cellular features are transparent, spatially subtle, or impossible to distinguish reliably without signal amplification or contrast enhancement. Fluorescence-based staining provides that contrast while also allowing multiple dimensions of cell biology to be examined in the same workflow. Researchers use cell staining to define morphology, identify intracellular compartments, compare treated and untreated samples, assess cell viability, build multicolor imaging panels, and connect structural information with functional readouts. In this sense, staining often serves as the interpretive foundation for the entire experiment. If the staining workflow is poorly matched to the sample, target, or platform, then every downstream conclusion becomes less secure.
This is also why troubleshooting cell staining requires more than simply asking whether the dye fluoresced. A stain can fluoresce strongly while still failing analytically. It may localize incorrectly, generate misleading background, overrepresent damaged cells, disappear after fixation, or interfere with other channels in a multiparameter panel. The central question is not whether fluorescence is present, but whether the observed fluorescence still corresponds to the intended biological feature under the exact conditions of the experiment. Once cell staining is understood in that deeper way, troubleshooting becomes more rational: the goal is not just to recover brightness, but to recover meaningful signal.
Why Cell Staining Problems Are Often Multi-Factorial?
One of the most common reasons cell staining troubleshooting becomes frustrating is that researchers often expect one clear failure point, when in reality most staining problems are produced by several interacting factors. A result that appears to reflect a simple issue such as weak signal or high background is often the combined consequence of sample condition, staining chemistry, incubation behavior, wash stringency, fixation effects, optical settings, and even how the experiment is interpreted after acquisition. This multi-factorial nature is especially important in fluorescence workflows because the final image or signal profile does not show the origin of the problem directly. It shows the accumulated outcome of the entire workflow. This means the visible failure is often only the surface expression of a deeper mismatch within the experimental design. A weak signal, for example, may be attributed to insufficient dye concentration, yet the real cause may be that the target was poorly accessible, the cells were unhealthy, the incubation conditions were suboptimal, and the signal was partially bleached during acquisition. Likewise, high background may not come only from excess dye. It may also reflect sample autofluorescence, poor compatibility between the dye and the buffer system, inadequate washing, or a fixation step that changed how the fluorophore behaved in the sample. When troubleshooting ignores this layered complexity, the experiment often enters a cycle of random adjustments that consume time without actually improving interpretability.
Why a Failed Staining Result Is Rarely Caused by One Factor Alone?
In most practical workflows, cell staining failure emerges from the interaction of multiple variables rather than a single isolated mistake. Cells may already be stressed before staining begins, which changes membrane permeability or uptake behavior. The dye may be technically suitable in general but poorly matched to the actual sample condition. Wash steps may be too gentle to remove free dye or too harsh to preserve weakly retained signal. The instrument settings may then amplify the problem further by saturating background or accelerating photobleaching. By the time the researcher sees the final result, these factors are layered together. This is why one-variable assumptions are often misleading. A bright background is not always just a washing problem, and a weak signal is not always just a concentration problem.
How Sample Condition, Dye Choice, Protocol Steps, and Imaging Setup Interact?
The interaction between these variables is what makes staining problems so difficult to diagnose casually. Sample condition influences how cells tolerate the stain, how they take it up, and whether damaged cells contribute misleading fluorescence. Dye choice influences specificity, brightness, retention, and compatibility with live or fixed processing. Protocol details such as incubation time, temperature, solvent exposure, and wash logic determine how the reagent actually behaves in the biological system rather than in the product description. Imaging or detection settings then decide how much of that behavior becomes visible, exaggerated, or masked. For example, a dye with acceptable signal-to-background performance may look unusable if the detector gain is set too high, while a genuinely poor staining pattern may look superficially acceptable if exposure is adjusted only to make the image attractive. This interconnectedness is why troubleshooting must evaluate the workflow as a system rather than as a list of isolated steps.
Why Troubleshooting Should Follow a Structured Sequence Rather Than Trial and Error?
Because staining failures are multi-factorial, the most effective troubleshooting strategy is sequential rather than reactive. Trial-and-error changes often fail because they alter several variables at once without revealing which variable actually mattered. If concentration, incubation time, and imaging settings are all changed together, an apparent improvement may be temporary or impossible to reproduce because the real source of the improvement remains unknown. A structured sequence works better: first assess whether the sample itself was suitable, then review whether the staining chemistry matches the biological question, then examine execution variables such as timing and washing, and finally assess optical detection and analysis assumptions. This order reflects the logic of the workflow itself. It begins with the biological substrate, moves through the staining mechanism, and ends with the readout system.
Need help improving staining consistency across cell types, assays, or imaging workflows?
We can help match staining strategies to your sample type, experimental setup, and detection method to reduce artifacts and improve reproducibility.
Problem 1: High Background Fluorescence
High background is one of the most common staining complaints because it reduces contrast, obscures weak targets, and makes localization harder to interpret. In cell staining, background is not always caused by one obvious mistake. It can arise from intrinsic sample fluorescence, excess free dye, non-specific interactions, or protocol conditions that encourage diffuse fluorescence rather than target-restricted signal.
- Autofluorescence from Cells, Media, or Sample Preparation: Some samples naturally emit fluorescence, especially in shorter wavelength channels, and media components, fixation reagents, or stressed cells can increase that background further. This can be especially problematic when weak biological signals are assigned to autofluorescence-prone regions of the spectrum. Checking unstained controls early helps distinguish biological fluorescence from sample-derived background before the staining workflow is blamed incorrectly.
- Excess Dye Concentration and Incomplete Washing: Overconcentrated dye solutions and insufficient wash steps are common reasons for diffuse fluorescence. Free dye remaining around or within the sample can create haze, poorly defined structures, or broad population positivity that is mistaken for true staining. In many cases, improving background does not require a completely new reagent; it requires a better concentration window and wash sequence that removes excess dye without stripping the target-associated signal.
- Non-Specific Staining and Poor Reagent Compatibility: Background can also come from the way a dye interacts with the sample rather than from how much dye is present. Some fluorophores accumulate in unwanted regions because of membrane affinity, charge interactions, protein binding, or poor compatibility with the chosen buffer or sample preparation. This type of background is often harder to solve by washing alone and may require rethinking the stain class, buffer system, or whether the sample is better suited to a different fluorophore family.
- Practical Ways to Reduce Background Without Losing True Signal: The safest way to reduce background is to optimize one variable at a time while monitoring whether the target-associated signal remains interpretable. Useful adjustments often include lowering dye concentration, shortening incubation, refining wash conditions, using appropriate unstained controls, avoiding unnecessary light exposure, and checking whether the chosen channel is heavily affected by autofluorescence. The goal is not the darkest possible image, but the highest ratio of real signal to irrelevant fluorescence.
Problem 2: Weak, Dim, or Unstable Fluorescence Signal
Weak or unstable fluorescence is often more frustrating than high background because the experiment may look almost successful while still being too unreliable for analysis. In these cases, the challenge is to improve useful signal without increasing non-specific staining or losing biological meaning.
- Low Dye Loading or Insufficient Target Accessibility: A stain may produce weak fluorescence if it does not reach the target efficiently, if the target is poorly accessible under the chosen sample condition, or if the dye simply does not accumulate strongly enough for the platform being used. This is especially common when live-cell-compatible dyes are applied to difficult intracellular targets or when a fixed-cell workflow does not include conditions that adequately expose the target structure.
- Incorrect Incubation Time, Temperature, or Concentration: Signal strength is highly sensitive to incubation variables. Too little time or too low a concentration may produce incomplete staining, while the wrong temperature can affect uptake kinetics or compartment behavior. These factors should be optimized together rather than independently, because changing one often alters the effect of the others. A useful signal is not simply a brighter one, but a stronger signal that still reflects the intended staining logic.
- Photobleaching Before or During Image Acquisition: Some weak signals are not weak at staining completion; they become weak because the fluorophore bleaches before or during acquisition. This is particularly important in confocal imaging, time-lapse microscopy, or repeated scanning workflows. If the signal appears strong in early fields but fades rapidly, the main problem may be light exposure and acquisition settings rather than the stain itself.
- How to Improve Signal Strength While Maintaining Specificity: Improving signal should start with target access, incubation logic, and acquisition protection rather than automatically increasing dye concentration. In many cases, signal becomes more usable when the stain is better matched to the sample state, the incubation window is optimized, and acquisition settings are adjusted to reduce bleaching. Stronger signal is only valuable when it remains target-specific and reproducible.
Problem 3: Uneven Staining, Poor Reproducibility, or Inconsistent Results Between Samples
Another common complaint is inconsistency: one sample looks good, another looks weak, or the same protocol behaves differently across runs. This kind of variability often reflects differences in sample condition or workflow execution rather than a fundamentally bad stain.
- Differences in Cell Density, Confluency, or Sample Health: Cell density and health strongly influence staining outcome. Overcrowded cultures, overly sparse samples, stressed cells, or variable culture history can change uptake, retention, morphology, and background. If staining quality varies from run to run, sample condition should be checked first because even small differences in confluency or viability can shift the apparent performance of the stain.
- Uneven Reagent Exposure, Poor Mixing, or Inconsistent Wash Steps: Uneven addition of reagents, insufficient mixing, localized evaporation, or inconsistent wash timing can all produce visible staining gradients or well-to-well variability. These problems are especially common in multiwell formats and in workflows where one operator changes technique between runs. Standardizing how reagents are added and removed is often as important as optimizing the stain itself.
- Batch-to-Batch Variability in Dyes, Cells, or Handling: Reproducibility problems can also arise when different reagent lots, cell passages, or handling practices are introduced without documentation. In fluorescence workflows, even moderate variation in stock preparation, storage history, or sample passage number can alter signal quality enough to change interpretation. Batch awareness is therefore part of staining quality control, not just a record-keeping exercise.
- How to Standardize the Workflow for Better Reproducibility: Better reproducibility usually comes from reducing procedural variation. This includes controlling cell confluency windows, using consistent incubation times and wash conditions, documenting stock preparation and storage, standardizing acquisition settings, and comparing results only when controls behave similarly across runs. Reproducibility is improved not by making the protocol more complicated, but by making it more consistent.
Problem 4: Spectral Overlap, Channel Interference, and False Co-Localization
As cell staining experiments become more complex, optical interference becomes a frequent source of misinterpretation. Multicolor workflows are particularly vulnerable because good-looking images can still contain strong spectral artifacts that distort the biology being inferred.
- Why Multiple Fluorescent Channels Can Interfere with Each Other: Fluorophores do not emit at one perfectly isolated wavelength. Their spectra extend across ranges, and those ranges can overlap with neighboring channels. When several dyes are combined, one signal may bleed into another detector window, making the channels less distinct than expected. This becomes especially problematic when the panel includes very bright stains alongside weak targets.
- Spectral Overlap vs Bleed-Through vs True Co-Localization: These terms are often confused, but they are not identical. Spectral overlap describes the optical property that makes interference possible, bleed-through describes the practical appearance of signal in the wrong channel, and true co-localization refers to genuine spatial overlap of two biological targets. Distinguishing between them is critical because false co-localization is one of the most common interpretive mistakes in multicolor staining.
- Compensation, Filter Choice, and Multicolor Panel Planning: Compensation or spectral separation tools can help manage overlap, but they do not replace good panel design. Choosing cleaner filter sets, assigning fluorophores according to target abundance, and planning channels early are usually more effective than relying on correction after the data are collected. This is why multicolor troubleshooting often leads back to multicolor cell staining design decisions rather than just to acquisition settings.
- How to Redesign a Multicolor Staining Workflow More Safely: If a multicolor panel repeatedly produces overlap-related ambiguity, the most robust fix is often redesign rather than further compensation. Useful redesign strategies include moving a weak target to a cleaner channel, separating co-expressed markers more aggressively, replacing highly overlapping fluorophores, and revalidating the panel with single-stain and unstained controls before repeating the main experiment.
Problem 5: Signal Changes Caused by Fixation, Permeabilization, or Sample Processing
Many researchers discover problems only after the staining step appears successful but the signal changes during processing. Fixation, permeabilization, and related treatments are not neutral operations. They can preserve useful information, but they can also change what the fluorescence signal actually means.
- Why Some Live-Cell Stains Lose Meaning After Fixation: Many live-cell dyes depend on active physiological conditions such as membrane integrity, organelle potential, or compartment-specific chemistry. Once the sample is fixed, those conditions may no longer exist, even if the fluorescence remains visible. In other cases, the signal may simply redistribute or fade. This is why a live-cell stain should never be assumed to remain biologically equivalent after fixation unless the workflow is known to support that transition.
- How Fixation Can Alter Localization, Brightness, or Background: Fixation may preserve structure, but it can also change molecular accessibility, concentrate fluorescence in unexpected regions, increase background, or alter how the dye partitions in the sample. A signal that looked clean before fixation may appear brighter, dimmer, or more diffuse afterward, not because the biology changed, but because the sample chemistry changed.
- Permeabilization-Related Extraction or Redistribution Artifacts: Permeabilization can improve access to intracellular targets, but it may also extract weakly retained molecules or redistribute loosely associated dyes. This is especially important in samples where the fluorescence signal depends on compartment retention rather than covalent or strongly stabilized association. A processing step that improves one target may therefore distort another.
- How to Judge Whether the Signal Change Is Technical or Biological: The safest way to distinguish technical artifacts from genuine biology is to compare the signal across defined workflow variants rather than relying on visual impression alone. If the signal changes only after a processing step while untreated biological controls remain otherwise similar, the change is more likely technical. This is one reason why the distinction explored in live cell staining vs fixed cell staining should be considered at the design stage rather than after problems appear.
Troubleshooting weak signal, high background, or uneven cell staining?
We can help you identify common staining issues and optimize probe selection, incubation conditions, and wash steps for cleaner, more reliable results.
A Practical Troubleshooting Workflow Before Repeating the Experiment
Once a staining result has failed, the instinct is often to repeat the experiment quickly with one obvious adjustment, such as increasing dye concentration, extending incubation time, or changing the exposure setting. In many cases, this approach leads to a second unsatisfactory run because it addresses the visible symptom rather than the actual source of the problem. A more useful strategy is to follow a structured troubleshooting workflow that reviews the experiment in the same sequence in which the staining result was generated. This allows researchers to identify whether the failure began with the sample, with the choice of stain, with the way the protocol was executed, or with the way the signal was collected and interpreted. A practical troubleshooting workflow is especially valuable because it helps distinguish between problems that require small optimization and problems that require a deeper redesign. Some failures can be corrected with tighter washing, improved control design, or more appropriate acquisition settings. Others reveal that the stain is fundamentally mismatched to the target, the sample state, or the platform. Repeating the same workflow without this distinction often increases time loss and decreases confidence in the data. By reviewing the experiment step by step, researchers can make a more rational decision about whether to optimize, replace, or restructure the staining strategy before committing to another full run.
- Step 1: Check Sample Condition First: The first step is to evaluate whether the sample itself was suitable before staining began. Cell health, density, confluency, attachment quality, handling stress, and media history can all affect staining outcome before the dye is even introduced. Poor sample condition can increase membrane permeability, alter uptake patterns, raise background, or create inconsistent morphology that later appears to be a staining problem. Because of this, troubleshooting should always begin by asking whether the cells were biologically and physically stable enough for the intended assay. If the sample was already compromised, optimizing the stain alone may not restore a meaningful result.
- Step 2: Review Dye Choice and Staining Logic: After sample condition, the next question is whether the chosen dye was actually appropriate for the biological target and workflow design. A structurally oriented stain may have been used where a functional readout was needed, a live-cell dye may have been expected to retain meaning after fixation, or a fluorophore may have been placed into a channel that made interpretation unnecessarily difficult. At this stage, researchers should reassess what the dye is designed to report, whether that matches the real experimental question, and whether its behavior is compatible with live-cell analysis, fixed-cell processing, multicolor use, or the intended platform. If the dye is conceptually mismatched, protocol refinement alone will usually not solve the problem.
- Step 3: Reassess Protocol Details and Wash Conditions: Once sample quality and stain suitability have been considered, the protocol itself should be reviewed carefully. This includes staining concentration, incubation duration, temperature, buffer composition, solvent exposure, mixing consistency, wash sequence, and light protection during handling. Small procedural differences often have larger effects than expected, especially in fluorescence-based workflows where background and signal retention are sensitive to physical conditions. Reassessing the protocol step by step helps determine whether the experiment failed because the stain was underloaded, overstained, poorly washed, unevenly applied, or destabilized during routine handling rather than because the reagent was inherently unusable.
- Step 4: Check Acquisition Settings and Controls: If the stain and protocol appear fundamentally reasonable, the next step is to examine how the signal was collected and interpreted. Weak signal can be exaggerated by low detector sensitivity or premature bleaching, while high background can appear worse under aggressive gain or exposure settings. In multicolor workflows, channel interference may be mistaken for biology if appropriate controls are missing. This is why unstained controls, single-stain controls, and consistent acquisition settings are essential parts of troubleshooting. They help reveal whether the problem lies in the staining result itself or in the way the readout system displays and separates the fluorescence.
- Step 5: Decide Whether to Optimize, Redesign, or Switch Strategy: After reviewing the sample, stain choice, protocol execution, and acquisition conditions, the final step is to decide what kind of correction the experiment actually requires. If the underlying design is sound, a limited optimization such as titration adjustment, gentler washing, better control design, or improved acquisition parameters may be enough. If the workflow shows deeper incompatibility, such as a stain that loses meaning after processing or a panel with unavoidable optical conflict, the better decision may be to redesign the protocol or switch to a different stain class entirely. Making this distinction before repeating the experiment is often what separates productive troubleshooting from repeated procedural drift.
Applications of Cell Staining in Research Workflows
Although cell staining is often introduced as a visualization method, its real value lies in how it supports different layers of biological interpretation across research workflows. A staining strategy can be used to define overall cell morphology, identify subcellular compartments, distinguish live and damaged cells, compare treatment responses, or integrate several readout types into one experimental system. This makes cell staining more than a technical add-on. In many fluorescence-based studies, it functions as the bridge between raw biological material and interpretable analytical data. The importance of application context is also directly relevant to troubleshooting. A staining pattern that is acceptable for simple morphology screening may be inadequate for quantitative localization work. A moderate level of background that can be tolerated in a broad structural image may be unacceptable in a viability assay or multicolor panel. For this reason, understanding the intended application of cell staining helps researchers judge whether a staining problem is minor, moderate, or fundamentally experiment-limiting. The same dye can behave very differently in value depending on what the workflow is trying to achieve.
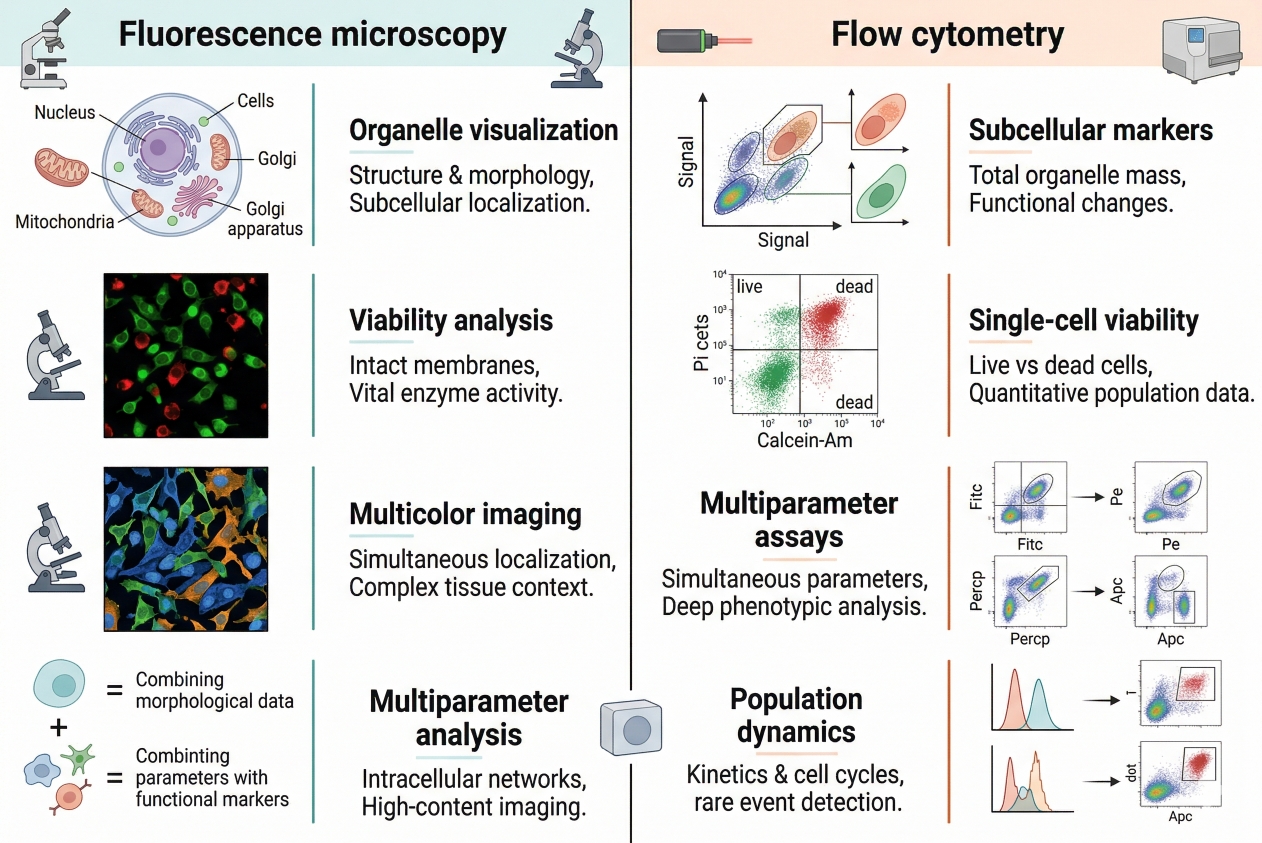 Fig. 2. Cell staining supports fluorescence microscopy, flow cytometry, organelle analysis, viability assessment, and multicolor research workflows (BOC Sciences Authorized).
Fig. 2. Cell staining supports fluorescence microscopy, flow cytometry, organelle analysis, viability assessment, and multicolor research workflows (BOC Sciences Authorized).
Cell Staining in Fluorescence Microscopy and Cell Imaging
In fluorescence microscopy and cell imaging, staining is most often used to create visual contrast that supports morphological analysis, intracellular localization, image segmentation, and structural interpretation. Nuclear stains can define cell number and spatial organization, membrane stains can clarify boundaries, and organelle-targeted probes can reveal how subcellular structures are distributed within the same field. In this context, the quality of cell staining is judged not only by brightness, but by localization fidelity, background control, and how clearly the signal preserves spatial meaning. This is why microscopy-oriented staining workflows are especially sensitive to false co-localization, photobleaching, diffuse background, and sample-processing artifacts.
Cell Staining in Flow Cytometry and Population Analysis
In population-based fluorescence workflows such as flow cytometry, cell staining is used less for spatial interpretation and more for discriminating subsets, defining fluorescence-positive events, excluding non-viable cells, and improving multiparameter analysis. Here, staining must support clean signal separation across large numbers of cells, often under conditions where small changes in background or overlap can alter gating logic and population boundaries. A stain that performs well in microscopy may not behave as well in flow if it lacks sufficient separation or creates broad distributions. For this reason, the usefulness of cell staining in population analysis depends heavily on reproducibility, signal consistency, and compatibility with multicolor panel design rather than on visual appearance alone.
Cell Staining in Organelle Visualization and Localization Studies
A major application of cell staining is the visualization of specific intracellular structures and compartments. Researchers frequently stain nuclei, membranes, mitochondria, lysosomes, cytoskeletal elements, lipid-rich regions, and other organelle-associated targets to study localization, redistribution, and structural changes across conditions. These workflows are especially valuable when biological meaning depends on where a signal appears inside the cell rather than simply whether fluorescence is present. Because localization studies are highly dependent on staining specificity, they are also especially vulnerable to misleading results when the dye accumulates non-specifically, redistributes after fixation, or changes behavior during sample processing. This is why organelle-focused staining often demands tighter control over protocol conditions than more general contrast-based workflows.
Cell Staining in Viability Assessment, Cell Tracking, and Multicolor Assays
Cell staining is also central in workflows that go beyond static structural imaging, including viability assessment, cell tracking, and multicolor assay design. In viability analysis, stains help distinguish intact cells from damaged or dead cells so that the rest of the experiment can be interpreted more reliably. In tracking studies, fluorescent labels make it possible to follow cells over time, assess movement, or observe signal dilution across biological change. In multicolor workflows, staining becomes a system-level tool that combines several readout layers within one sample, such as structure, function, localization, and cell state. These applications place especially high demands on stain compatibility because the signal must remain meaningful not only in isolation, but also across time, across channels, or through multiple stages of experimental handling.
How BOC Sciences Supports Cell Staining Needs?
BOC Sciences supports cell staining needs through integrated capabilities in fluorescent dye and probe supply, custom probe development, multicolor staining strategy design, and workflow-oriented application support. Rather than treating cell staining as a simple reagent choice, we help researchers align staining targets, fluorophore properties, imaging conditions, and downstream analysis goals with the real demands of the experiment. This is especially important in cell staining workflows, where organelle specificity, live-cell compatibility, signal balance, and platform fit all influence whether the final staining result is truly useful. Our support is therefore structured to cover both routine cell visualization needs and more advanced imaging, screening, and multiparameter analysis applications.
Fluorescent Dye and Probe Supply for Cell Staining
- Access to a broad range of cell staining fluorescent dyes and probes covering multiple cellular structures, including nuclei, mitochondria, endoplasmic reticulum, cell membranes, and cytoskeletal components.
- Support for selecting fluorophores across multiple spectral regions, including blue, green, red, and near-infrared fluorescence, to better fit multichannel imaging workflows.
- Availability of diverse staining reagents suitable for both live-cell and fixed-cell applications, depending on sample handling strategy and imaging goals.
- Product support for highly selective, high signal-to-noise fluorescent probes that can be adapted to different experimental systems, microscopy platforms, and assay formats.
Customized Cell Staining Probe Development
- Custom design of fluorescent probes targeted to specific organelles, cellular structures, or function-related staining objectives according to individual research needs.
- Development support for functional probe systems used in fluorescence-based detection of ROS, pH variation, ion concentration, metabolic state, and related intracellular readouts.
- Structural optimization of fluorescent dyes to improve cell permeability, targeting selectivity, signal stability, and lower-disturbance staining performance.
- One-stop development services covering molecular design, synthesis, modification, purification, and characterization for customized cell staining probes.
Multicolor Staining Strategy Design and Application Support
- Practical support for designing multicolor cell staining workflows that improve compatibility between channels, balance fluorescence intensity, and reduce avoidable spectral interference.
- Application guidance for more complex experimental systems, including multi-organelle co-staining, combined structural and functional imaging, and broader panel-based cell analysis.
- Suggestions for optimizing staining conditions such as dye concentration, incubation time, buffer system, washing strategy, and imaging timing according to the actual assay context.
- Troubleshooting assistance for common staining challenges, including channel crosstalk, high background, photobleaching, weak localization contrast, and reduced interpretability in multicolor imaging.
Integrated Fluorescence Workflow and High-Content Application Support
- Support for integrating cell staining into broader experimental workflows that connect staining, imaging, analysis, and screening rather than treating each step in isolation.
- Guidance for staining strategies compatible with high-content screening (HCS) and high-throughput screening (HTS), helping researchers build more scalable fluorescence assay workflows.
- Application support for image-based phenotypic analysis, image segmentation, and multiparameter fluorescence readout design where staining quality directly affects downstream analytical performance.
- Workflow-oriented assistance aimed at improving experimental consistency, data comparability, and cross-platform interpretability in larger cell imaging and screening programs.
Do You Need A Consultation?
BOC Sciences integrates cutting-edge fluorescence technologies to accelerate your research, driving next-generation solutions for drug discovery and diagnostics.
Transform Your Studies with Cutting-Edge Fluorescent Products
| Catalog | Name | CAS | Inquiry |
|---|---|---|---|
| A16-0086 | 1,6-Diphenyl-1,3,5-hexatriene | 1720-32-7 | Bulk Inquiry |
| A16-0055 | NBD Sphingosine | 1449370-25-5 | Bulk Inquiry |
| A16-0016 | 5-Carboxyfluorescein N-succinimidyl ester | 92557-80-7 | Bulk Inquiry |
| A16-0003 | Phalloidin-TFAX 488 | 289620-19-5 | Bulk Inquiry |
| A16-0002 | Phalloidin-TRITC | 915013-10-4 | Bulk Inquiry |
| A16-0024 | Green CMFDA | 136832-63-8 | Bulk Inquiry |
| A16-0184 | 8-Methoxypyrene-1,3,6-trisulfonic acid trisodium salt | 82962-86-5 | Bulk Inquiry |
| A16-0157 | 3,3'-Dioctadecyloxacarbocyanine perchlorate | 34215-57-1 | Bulk Inquiry |
| A16-0166 | 3,3'-Diheptylthiacarbocyanine iodide | 53213-88-0 | Bulk Inquiry |
| A16-0151 | 3,3'-Diethylthiacarbocyanine iodide | 905-97-5 | Bulk Inquiry |
High-Performance Fluorescent Tools for Your Research
- Ion Fluorescent Probes Indicators for real-time ion concentration imaging.
- Mitochondrial Fluorescent Probes Targeted dyes for mitochondrial structure and function.
- Nerve Terminal Probes Fluorescent tracers for synaptic activity analysis.
- Metal Fluorescent Probes Selective sensors for intracellular metal ions.
- Cell membrane Fluorescent Probes Surface-labeling dyes for membrane dynamics studies.
- Lysosomal Fluorescent Probes Acidic organelle markers for lysosome tracking.
- Endoplasmic Reticulum Fluorescent Probes ER-targeted dyes for organelle structure analysis.
- Nitric Oxide (NO) & Reactive Oxygen Species (ROS) Probes for oxidative stress and signaling detection.
Explore More Topics
Online Inquiry